About PMM2-CDG (CDG-1a)
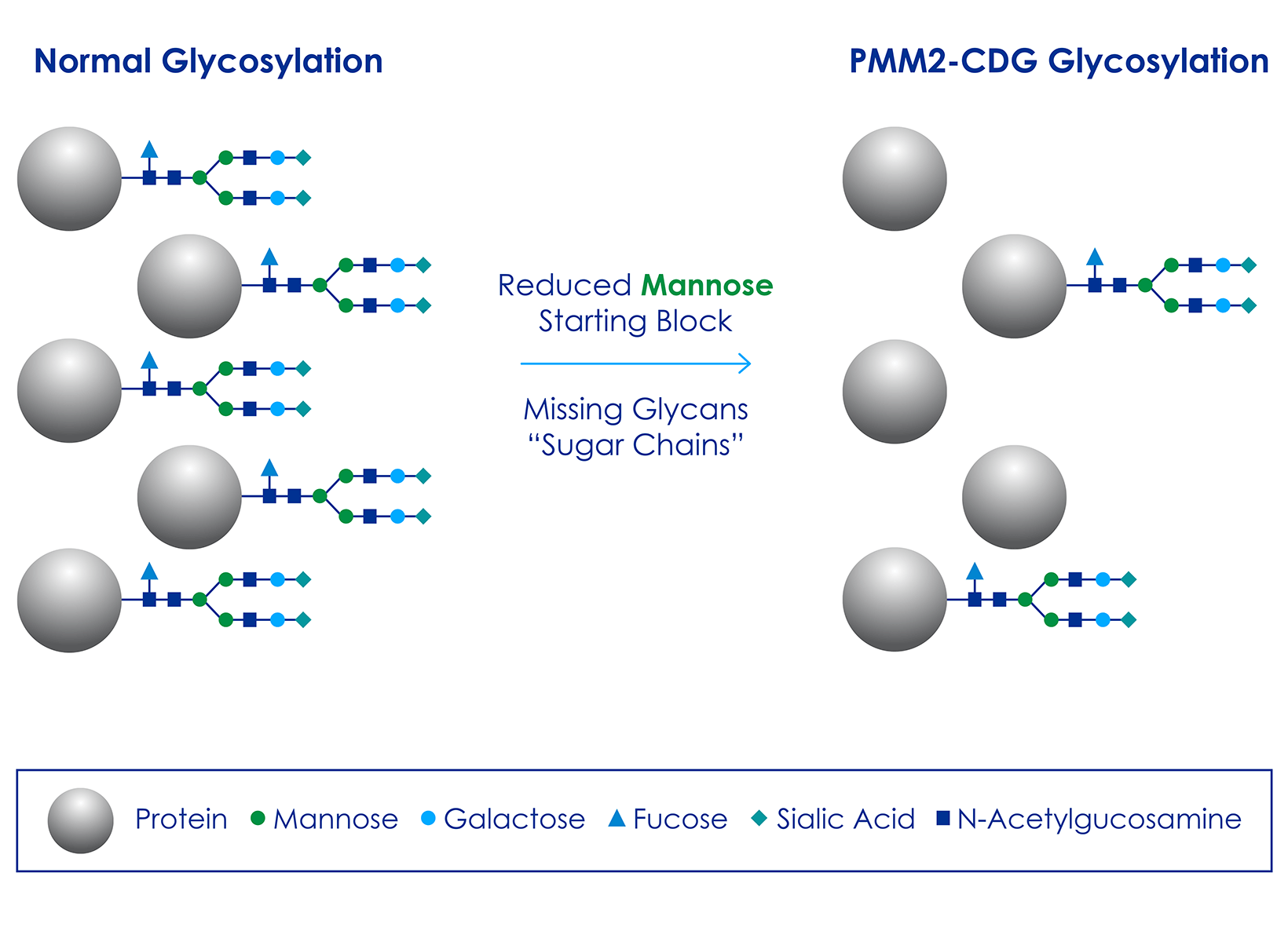
PMM2-CDG (also known as CDG-1a) is short for phosphomannomutase 2-congenital disorder of glycosylation and is the most prevalent congenital disease of glycosylation (CDG). Glycosylation is the process of adding sugar chains, also called glycans, to the surface of proteins. More than 50% of proteins in the body are glycosylated and this process is critically important to ensure the correct structure and function of proteins.