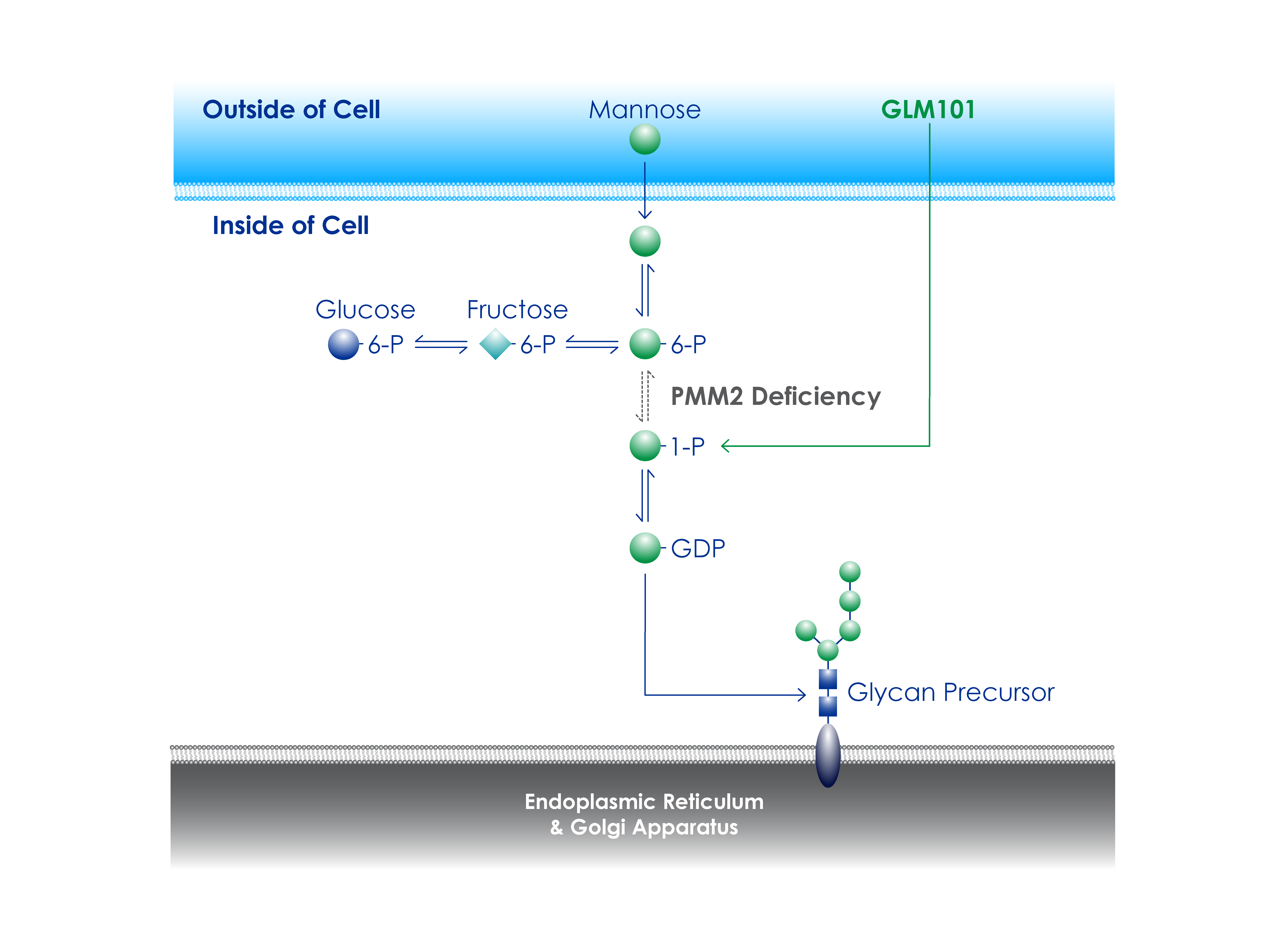
GLM101: a potential PMM2-CDG treatment
Glycomine is developing orphan drugs for serious rare disorders of metabolism and protein misfolding, where no other therapeutic options exist and replacing a missing component would restore function. The company’s replacement therapy approach aims to deliver substrates, enzymes, or proteins and to target these molecules to clinically relevant cellular compartments. GLM101 is Glycomine’s first drug from its platform and is designed to intracellularly deliver mannose-1-phosphate as a potential treatment for PMM2-CDG (also known as CDG-1a). GLM101 is currently in clinical trials in the U.S. and Europe and has received Orphan Drug Designation (ODD) in the U.S. and Europe and Rare Pediatric Disease Designation (RPDD) in the U.S.
View our Policy on Expanded Access here.